












CRISPR-based therapies offer transformative potential for treating previously untreatable conditions, particularly rare genetic diseases. However, navigating the regulatory approval process remains a significant challenge. This article outlines the complex development pathway from initial discovery through FDA review and emphasize the importance of strategic partnerships and adherence to current good manufacturing practices (CGMP) to ensure these innovative CRISPR-based therapies are safe and effective for patients.
CRISPR-based therapy development: key regulatory milestones from discovery to FDA approval
Developing CRISPR-based therapies from early-stage research to clinical application is a rigorous, multi-phase process requiring strict regulatory oversight (Table 1). Drug discovery and development are divided into various stages. Once preclinical research is completed and approved by regulatory agencies, the next stage in the development is conducting human trials. These trials occur in three phases to evaluate the drug’s safety, efficacy, and side effects, followed by large-scales testing prior to final approval for market use.
Table 1. Regulatory pathway for CRISPR-based therapies
| Step | Phase/Application | Description |
| Step 1 | Discovery phase | Development begins with identifying promising therapeutic targets and formulating hypotheses for further investigation. |
| Step 2 | Preclinical research | Before initiating human trials, CRISPR-based therapies undergo rigorous preclinical evaluation to assess safety and efficacy through both in vitro and in vivo studies. These studies form the foundation for submitting an Investigational New Drug (IND) application. |
| Step 3 | Clinical research (IND, Phase 1–3) | IND submissions are reviewed by the FDA, and upon approval, CRISPR-based therapies progress through Phases 1, 2, and 3 clinical trials. Each phase is designed to evaluate safety, efficacy, and optimal dosing in human subjects. |
| Step 4 | BLA Application | Upon successful completion of clinical trials, developers submit a Biologics License Application (BLA) to the FDA. The agency reviews all relevant data to determine approval status. |
| Post approval | Surveillance | Even after approval, CRISPR-based therapies remain subject to ongoing FDA monitoring to ensure long‑term safety and efficacy. |
The Investigational New Drug (IND)/Clinical Trial Application (CTA) and approval cycle
The complex journey of a CRISPR-based therapy involves a rigorous regulatory foundation. Once preclinical research is completed and approved by the regulatory agencies, the next stage in the development process is the Investigational New Drug (IND) application (Figure 1). The IND is the formal request to the FDA to begin human clinical trials and must contain comprehensive data from preclinical research (safety and efficacy in the lab or animals), detailed Chemistry, Manufacturing, and Controls (CMC) information demonstrating product quality and consistency [2]. The FDA's initial review of a full IND submission takes 30 calendar days, during which the agency determines if the therapy is safe to proceed to clinical trials. Unlike traditional small-molecule drugs, the complexity of CRISPR-based therapies often requires extensive pre-IND and early-phase interaction with the FDA's Center for Biologics Evaluation and Research (CBER) to align on manufacturing processes and long-term safety monitoring plans.
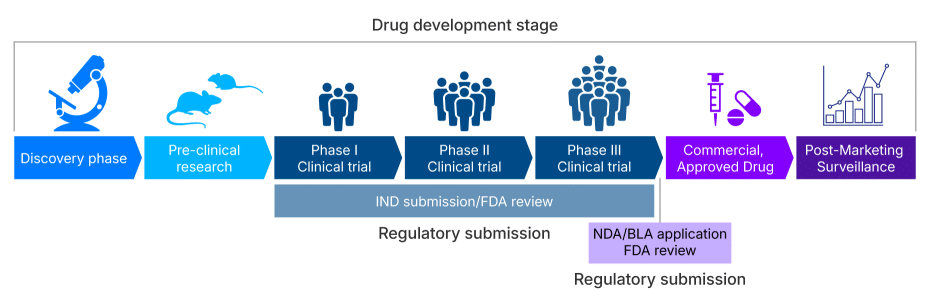
Similarly for the CGT developers targeting the European market, the equivalent milestone to the FDA's IND is the Clinical Trial Application (CTA). The CTA is submitted to the relevant national competent authorities under the framework of the European Medicines Agency (EMA) [3]. While the fundamental requirements for demonstrating product quality (CMC), non-clinical safety are similar to the IND, the CTA process introduces unique regional considerations. Navigating the differing submission procedures across European member states and understanding the centralized review process for certain Advanced Therapy Medicinal Products (ATMPs) is vital for achieving concurrent global clinical development [4].
Accelerating Cell and Gene Therapy (CGT) approvals
The FDA recognizes transformative potential and the urgent unmet medical need addressed by CGTs, particularly for rare diseases. To accelerate application approvals and meet the growing pipeline, the agency's Office of Therapeutic Products (OTP) has been strategically implemented with the expertise to review submissions. Furthermore, the FDA is actively issuing draft guidance documents to provide regulatory flexibility and clarity for CGT developers. The FDA also encourages the use of expedited programs such as Breakthrough Therapy (BT) and Regenerative Medicine Advanced Therapy (RMAT) designations, which provide intensive guidance and organizational commitment to expedite development and review [5]. This proactive approach aims to balance the need for rigorous safety and efficacy data with the goal of bringing life-changing therapies to patients.
The role of strategic partnerships: Leveraging expertise for success
Given the intricate and evolving regulatory environment, CGT developers are advised to seek specialized guidance. Strategic partnerships with entities experienced in navigating the regulatory landscape, such as Integrated DNA Technologies (IDT), can be invaluable. These partners offer expertise from the pre-clinical stages through IND submission and beyond, providing a roadmap for successful navigation through the regulatory process.
For businesses in the CGT domain, understanding the regulatory landscape is not merely a compliance requirement but a strategic advantage. The complexities of CGT development demand a proactive approach, where regulatory challenges are anticipated and addressed with precision and expertise. By strategic partnerships with IDT and leveraging the wealth of guidance available, CGT developers can navigate the regulatory maze more effectively, bringing life-changing therapies from concept to cure with greater efficiency and success. Contact us today for a free consultation on your CRISPR therapeutic development project and discover how our expertise can help you achieve your goals.
Decode the FDA and EMA requirements for CRISPR-based therapies with our regulatory eBook
Are you developing a groundbreaking CRISPR-based therapy? Don't let regulatory hurdles consume your timeline. While this article outlines the FDA pathway, our expert-written eBook, Navigating Regulatory Barriers for CRISPR-based Therapies, empowers you to confidently navigate the complex landscapes of both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).
This essential guide decodes the key regulatory elements required for developing and achieving FDA and EMA approval of CRISPR-based therapies:
- Understand Global Regulatory Frameworks: Review region-specific requirements for Investigational New Drug (IND) applications in the US and Clinical Trial Applications (CTAs) in the EU to ensure compliance with global standards.
- Address CMC and Quality Considerations: Ensure consistency and quality in CRISPR-based therapies by meeting FDA and EMA expectations for Chemistry, Manufacturing, and Controls (CMC).
- Ensure CGMP Compliance: Follow Current Good Manufacturing Practices (CGMP) and maintain full traceability in line with ICH Q7 guidelines to support your regulatory submissions.
- Navigate Regulatory Pathways: Gain comprehensive insight into the development journey of CRISPR-based therapies from early research through clinical application by understanding key regulatory milestones.
Download your copy of Navigating Regulatory Barriers for CRISPR-based Therapies today to master the regulatory requirements and accelerate your path to the clinic.