Ligation-based library preparation
Overview
To prepare samples for next generation sequencing (NGS), they must first be transformed into libraries—pools of DNA fragments with adapters that provide sequencing compatibility on a specific platform. Preparing quality libraries opens the door to discovery through a variety of NGS-based applications.

How to prepare a DNA Library
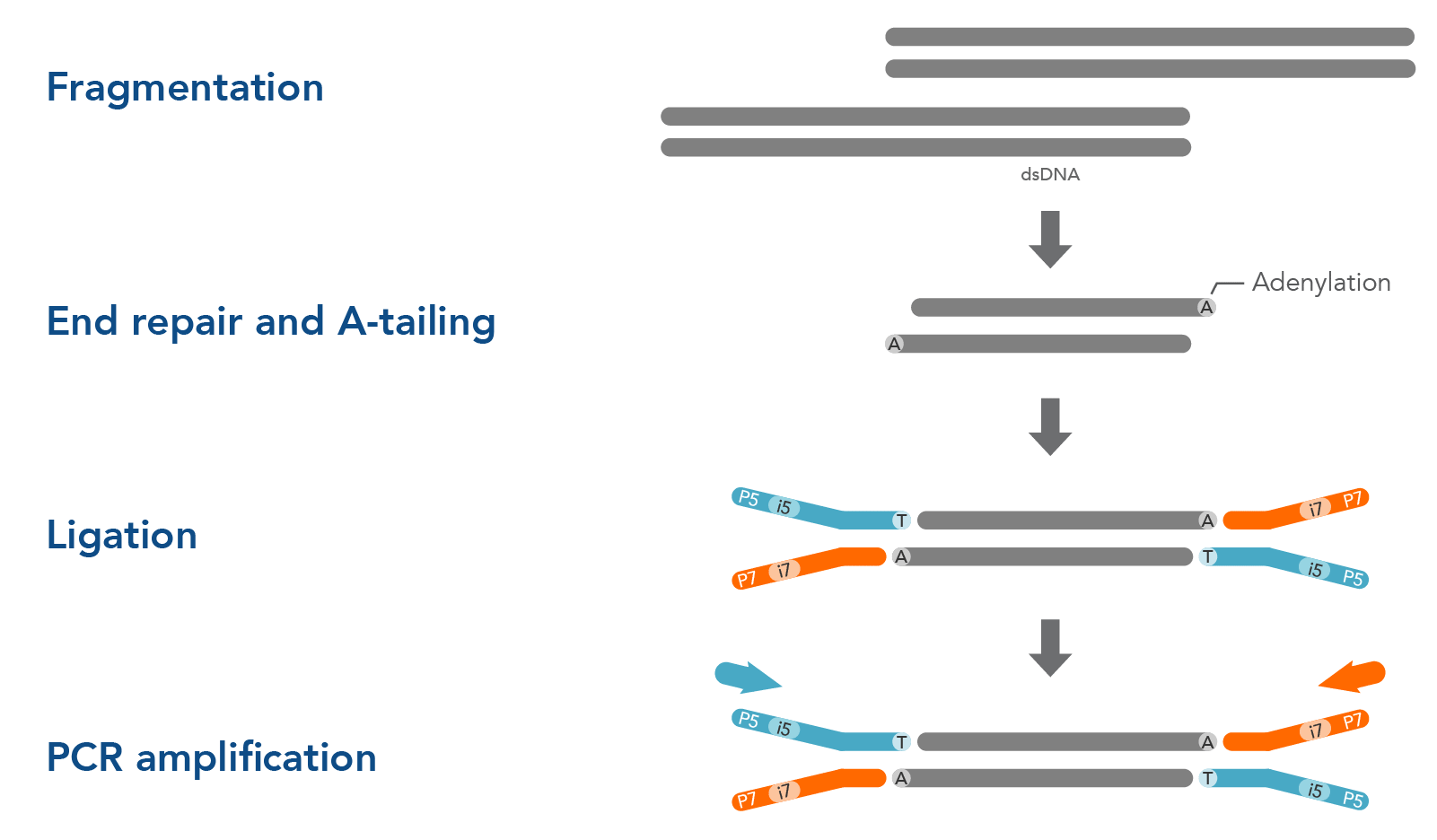
Before DNA or RNA samples can be sequenced, they must first be fragmented, end-repaired, and ligated to sequencing adapters. There are a variety of options for library preparation, and the precise protocol you use can influence your NGS sequencing results. Ligation-based library preparation is known for its high coverage uniformity, precise strand information, and reliability. The major steps of ligation-based library preparation are pictured in Figure 1 and summarized here:
- Fragmentation and end repair—Short-read sequencing technologies like those from Illumina™ cannot readily analyze very long DNA strands, so samples are fragmented into shorter, uniform pieces to prepare them for sequencing. Typically, whole genome sequencing (WGS) works best with 350 bp fragments, while hybridization capture works best with 200 bp fragments. After fragmentation, the DNA is end-repaired or end-polished. Generally, a single adenine base is added to form an overhang by an A-tailing reaction. This A overhang allows adapters containing a single thymine overhanging base to pair with the DNA fragments.
- Adapter ligation—A ligase enzyme covalently links the adapter to the DNA fragments, making a complete library molecule. Adapters serve multiple functions: they can attach the sequences to a flow cell or ensure sequencing platform-specific compatibility. They can contain barcodes—often called indexes—to identify samples and allow multiplexing in both target enrichment and sequencing. Adapters can also contain unique molecular identifiers (UMIs) which can help increase variant identification. Read more about Adapters for next generation sequencing.
- PCR amplification (optional)—Whether you amplify your libraries or not depends on the adapter type and input sample. After PCR amplification, free oligonucleotides and small fragments must be removed. PCR cleanup can be performed using magnetic beads, or a spin column.
Fragmentation Methods for Ligation-based library prep
There are two main DNA fragmentation methods for ligation-based library prep:
- Physical—When DNA is physically sheared using acoustics, nebulization, centrifugal force, needles, or hydrodynamics. While these methods achieve accurate, unbiased results, they require specialized machinery. For example, genomic DNA, or DNA derived from formalin-fixed paraffin-embedded (FFPE) samples, may be fragmented using Covaris® shearing. Use our xGen DNA Library Prep MC Kit (IDT) as it’s compatible with any physically fragmented DNA, including Covaris® sheared DNA.
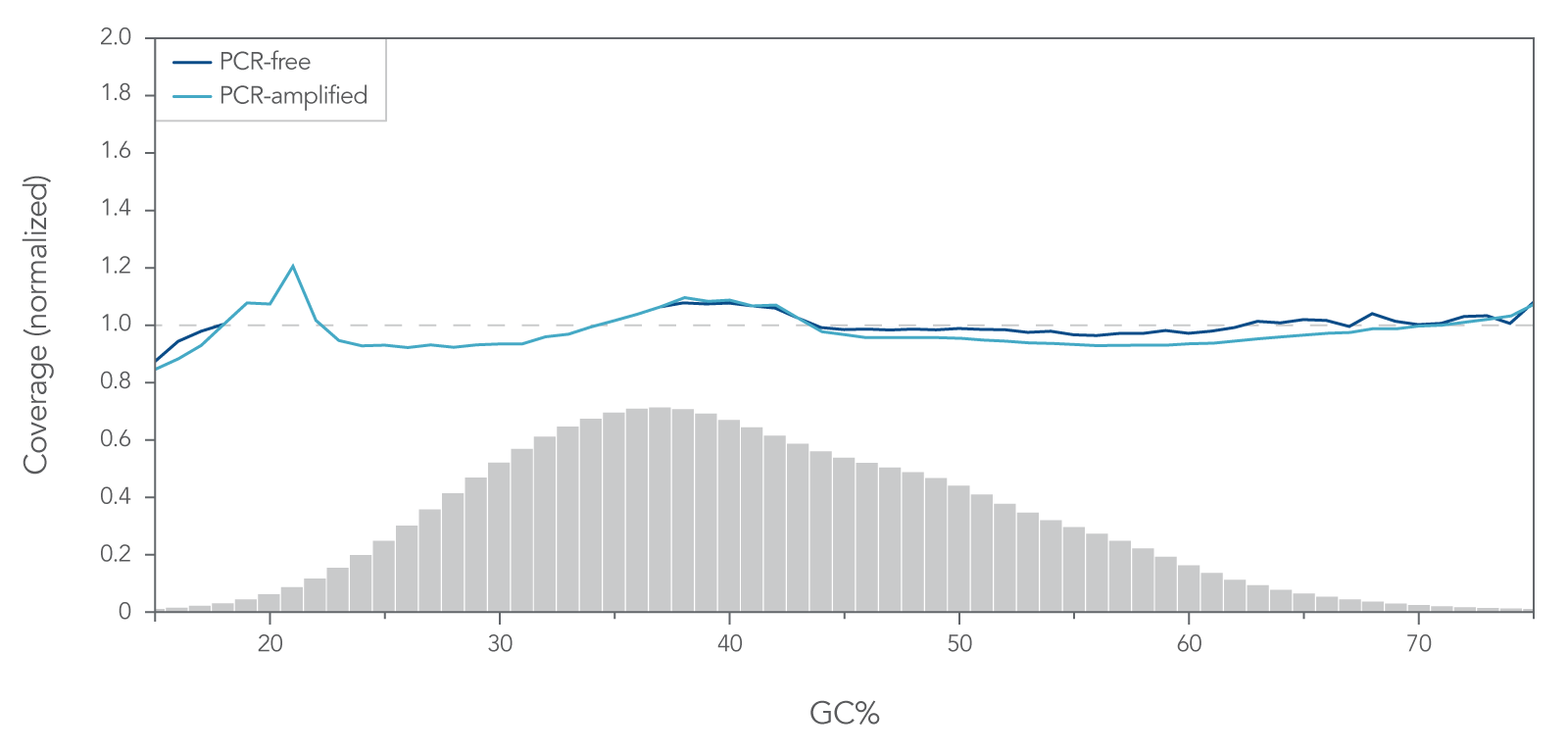
- Enzymatic— When DNA is digested into fragments using enzymes, and then undergoes end-repair and A-tailing. This method sometimes introduces a GC bias that does not occur during physical fragmentation; however, the method is quick, easy, and does not require any special equipment. IDT has optimized enzymatic DNA fragmentation to achieve consistent coverage without a GC bias in our xGen DNA Library Prep EZ Kit (Figure 2).
Some DNA does not require fragmentation because it is already within the size range required by the sequencer.
Downstream applications
- Whole genome sequencing (WGS)
- PCR-free, or PCR-amplified sequencing
- Identification of germline-inherited single nucleotide polymorphisms (SNPs), copy number variants (CNVs), and insertions/deletions (indels)
- Hybridization capture of target sequences (e.g., the exome or transcripts of interest)
- Low-frequency somatic variation identification of single nucleotide variants (SNVs), CNVs, and indels
- RNA-seq
- Metagenomic sequencing
Get started with library preparation solutions
Are you working in one of these applications, or about to get started? Take advantage of IDT’s expertise and specialized library prep kits to streamline your workflows and maximize your results.